专业科学仪器及设备制造商

DOI:10.1002/ece2.70093
全文速览
光催化氮还原合成氨是绿色、可持续的“零碳”路线,但N≡N键的高解离能(945 kJ/mol)和水中质子供给不足严重制约其效率。本研究在InVO₄纳米棒中通过快速焦耳热引入高密度氧空位(Vo),并利用紫外光诱导法将Ru纳米颗粒精准锚定于Vo位点,构建了Ru-V双位点协同催化体系。Ru位点负责水分子解离提供活性氢(*H),相邻的不饱和V位点负责N₂的吸附与活化,二者协同降低了反应能垒。最优催化剂Ru/InVO₄-Vo在纯水条件下氨产率达87.86 μmol·g⁻¹·h⁻¹,是单纯氧空位样品的2.3倍,且循环4次后活性保持>92%。原位XPS和DFT计算揭示了电子金属-载体相互作用(EMSI)驱动的定向电荷转移机制,以及“远端路径”加氢反应途径。该工作为光催化固氮的双位点协同设计提供了新思路。
背景介绍
氨是重要的化工原料和无碳能源载体,传统Haber-Bosch工艺高温高压、能耗巨大、碳排放高。光催化氮还原以水和氮气为原料,在常温常压下利用太阳能驱动,是理想的绿色合成氨路线。然而,N≡N三键极其稳定(945 kJ/mol),且光催化过程中活性氢(*H)的供给速率与N₂活化速率常不匹配,导致反应效率低下。
InVO₄作为一种可见光响应的窄带隙(~2.18 eV)半导体,具有稳定的晶格结构和本征的氧空位形成倾向,是光催化固氮的理想载体。然而,单纯氧空位改性仍面临水分子解离能力弱、缺乏特异性N₂吸附位点的瓶颈。近年来,Ru基材料因其适中的N₂吸附强度和优异的水解离能力而备受关注。本研究通过快速焦耳热引入氧空位+紫外光诱导锚定Ru纳米颗粒的策略,在InVO₄表面构建了Ru-V双位点协同体系,实现了高效N₂活化和*H供给的匹配。
本文亮点
(1)快速焦耳热引入氧空位:利用焦耳热(1000 ℃,H₂/Ar气氛)在InVO₄中引入高密度氧空位,XPS显示V⁴⁺/V⁵⁺比达0.64,EPR证实氧空位信号显著增强。
(2)紫外光诱导精准锚定Ru:Ru纳米颗粒(~2 nm)通过紫外光还原选择性锚定于氧空位位点,形成稳定的Ru-V配位结构。XPS证实Ru⁰占比约70%,HRTEM观察到Ru(101)晶格条纹(0.21 nm)。
(3)双位点协同催化:Ru位点解离水提供*H,不饱和V位点吸附活化N₂,DFT计算表明Ru-V桥位N₂吸附能仅0.03 eV,远低于单Ru位点。反应遵循“远端路径”加氢机制。
(4)优异光催化性能:纯水条件下氨产率87.86 μmol·g⁻¹·h⁻¹,为单纯氧空位样品的2.3倍;AQE达0.21%(365 nm);循环4次活性保持>92%。
图文解析

图1:催化剂合成路线与形貌表征
(a) Ru/InVO₄-Vo的设计合成路线;(b–c) InVO₄-Vo和Ru/InVO₄-Vo的SEM图像;(d) TEM图像;(e) HRTEM图像;(f) EDS面分布(In蓝、O紫、V绿、Ru红)。
图1a展示三步法:水热合成InVO₄纳米棒→焦耳热引入氧空位→紫外光诱导锚定Ru。SEM(图1b,c)和TEM(图1d)表明处理后纳米棒形貌保持完整,结构稳定。HRTEM(图1e)中0.273 nm对应InVO₄(112)面,0.21 nm对应金属Ru(101)面,证实Ru以金属态纳米颗粒形式锚定。EDS(图1f)显示Ru均匀分散,无团聚。
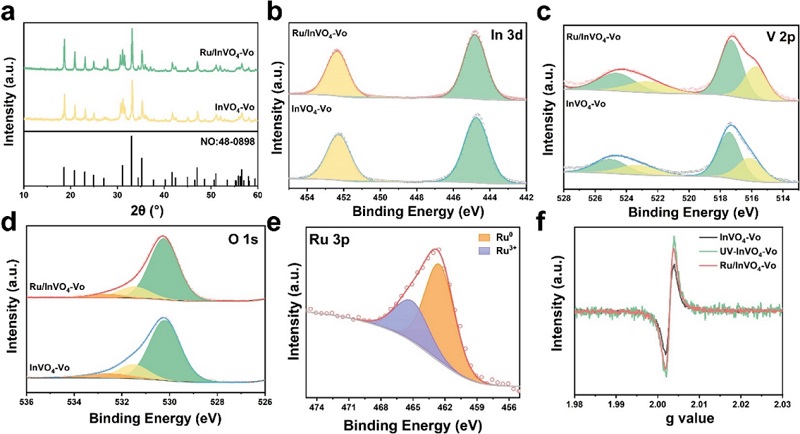
图2:物相与电子结构表征
(a)XRD谱图;(b) In 3d、(c) V 2p、(d) O 1s XPS谱图;(e) Ru 3p XPS谱图;(f) EPR谱图。
XRD(图2a)显示所有样品均与InVO₄标准卡片(PDF#48-0898)吻合,无Ru相关衍射峰,表明Ru高度分散。XPS(图2b–d)中Ru负载后In和O的结合能正移、V的结合能负移,表明电子从InVO₄向Ru转移,同时反馈至相邻V位点。V⁴⁺/V⁵⁺比为0.64证实氧空位存在;O 1s中表面氧空位含量从19.1%降至16.4%,表明Ru优先占据Vo位点。Ru 3p(图2e)中Ru⁰占比~70%,证实UV诱导成功制备金属态Ru。EPR(图2f)中g=2.003信号显示Ru/InVO₄-Vo的EPR强度介于InVO₄和UV-InVO₄-Vo之间,证实Ru部分占据氧空位。
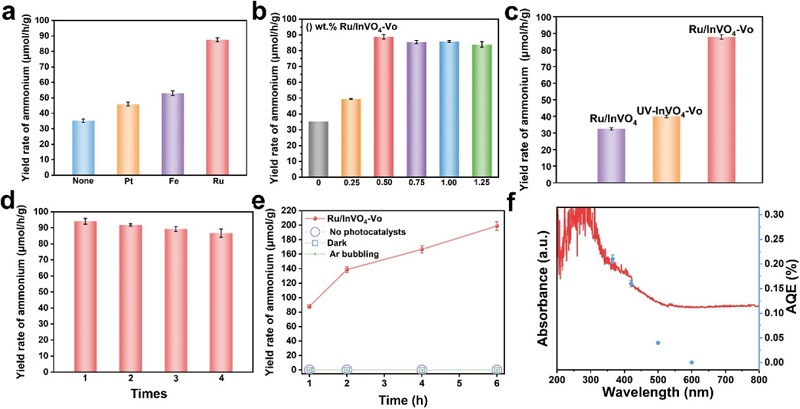
图3:光催化固氮性能
(a)不同金属(Ru、Pt、Fe)负载InVO₄-Vo的固氮性能;(b) 不同Ru含量的影响;(c) 不同InVO₄基催化剂的性能对比;(d) 循环稳定性测试;(e) 长期稳定性测试(6 h);(f) AQE(蓝点)与紫外-可见吸收光谱(红线)。
随S图3a中Ru/InVO₄-Vo活性最高(87.86 μmol·g⁻¹·h⁻¹),显著优于Pt和Fe体系,归因于Ru适中的N₂吸附强度和优异的水解离能力。图3b显示0.5 wt% Ru为最优载量。图3c中Ru/InVO₄-Vo的活性为单纯氧空位(UV-InVO₄-Vo)的2.3倍,证实性能提升源于Ru-V双位点协同而非单纯氧空位增加。图3d显示4次循环后保持>92%活性。图3e显示6 h累积NH₃产量达198.85 μmol·g⁻¹,呈线性增长。图3f中AQE分布与UV-vis吸收光谱吻合,365 nm处AQE达0.21%。
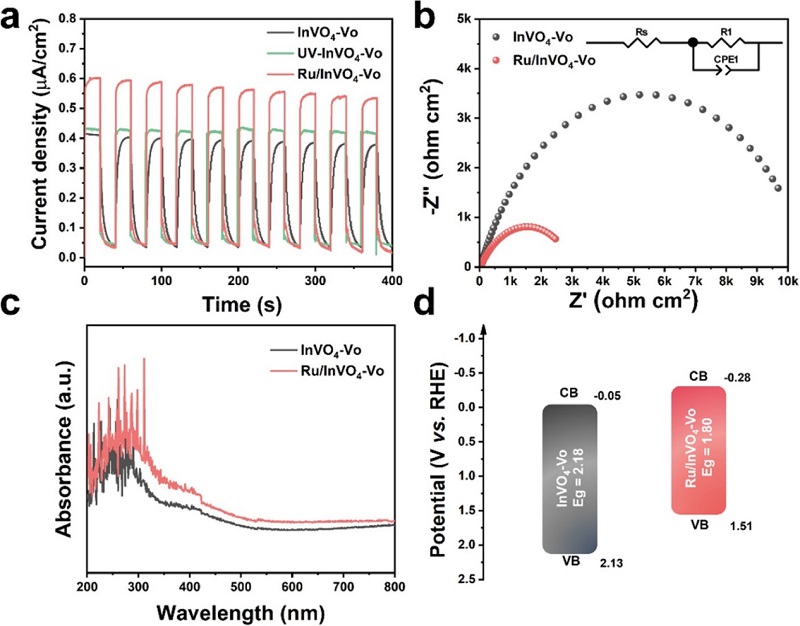
图4:光电化学性质与能带结构
(a) InVO₄-Vo、UV-InVO₄-Vo和Ru/InVO₄-Vo的光电流响应;(b) EIS Nyquist图;(c) DRS光谱;(d) InVO₄-Vo和Ru/InVO₄-Vo的能带结构示意图。
光电流(图4a)显示Ru/InVO₄-Vo的电流密度最高,证实Ru负载有效抑制了光生载流子复合。EIS(图4b)中Ru/InVO₄-Vo的圆弧半径最小,电荷转移电阻最低,表明界面电荷传输更快。DRS(图4c)显示Ru负载后光吸收增强,带隙从2.18 eV降至1.80 eV。能带结构(图4d)表明Ru/InVO₄-Vo的导带底(–0.28 eV)低于NRR热力学电位(–0.05 V),具备驱动固氮反应的热力学条件。
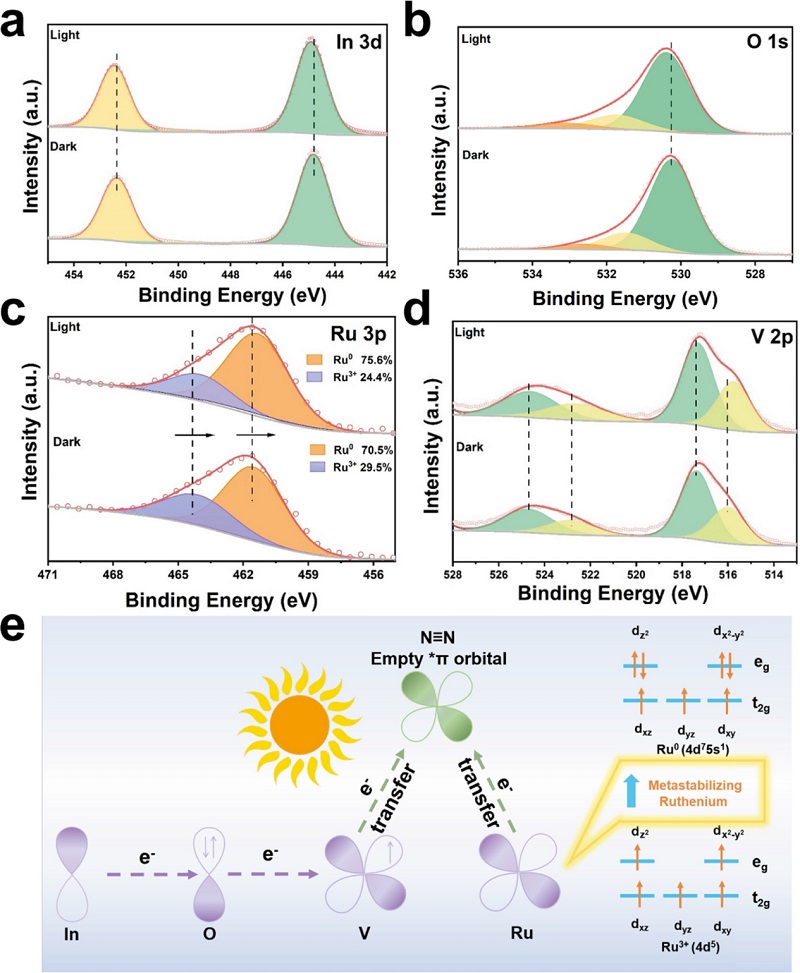
图5:原位XPS与电荷转移机制
Ru/InVO₄-Vo在光照前后的(a) In 3d、(b) O 1s、(c) Ru 3p、(d) V 2p的原位XPS谱图;(e) 光催化NRR过程中Ru/InVO₄-Vo的电荷转移示意图。
光照下In 3d和O 1s向高结合能偏移(电子密度降低),V 2p和Ru 3p向低结合能偏移(电子密度增加),证实光生电子选择性富集于V和Ru位点。Ru⁰占比从70.5%升至75.6%,表明Ru在光照下处于更富电子态。光照后各峰位恢复,证实结构可逆。图5e示意了电荷转移路径:光生电子从InVO₄向Ru迁移,富电子Ru反馈电子至相邻不饱和V位点,形成稳定的Ru-V配位结构,协同活化N₂。
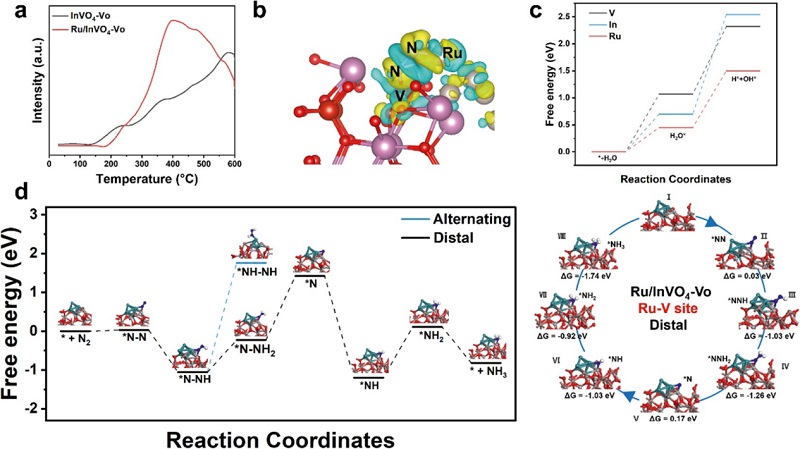
图6:N₂吸附、DFT计算与反应路径
(a) InVO₄-Vo和Ru/InVO₄-Vo的N₂-TPD曲线;(b) 差分电荷密度图;(c) Ru/InVO₄-Vo上HER的吉布斯自由能图;(d) 远端和交替机制下NRR的自由能图及对应反应能。
N₂-TPD(图6a)显示Ru/InVO₄-Vo在300–500 ℃出现显著N₂化学脱附峰,证实Ru负载增强了N₂化学吸附。差分电荷密度(图6b)显示N₂分子周围黄色(电子积累)区域显著,Ru-V位点周围蓝色(电子耗尽),证明双位点协同向N₂π轨道注入电子。HER自由能(图6c)表明Ru位点具有适中的H吸附能,可高效解离水提供H。NRR自由能(图6d)显示远端路径能垒(0.83 eV)远低于交替路径(2.82 eV),证实反应遵循“远端加氢”机制:N₂→NNH→NNH₂→N→NH→NH₂→NH₃。
总结与展望
本研究通过快速焦耳热引入氧空位结合紫外光诱导锚定,成功构建了Ru/InVO₄-Vo双位点光催化固氮体系。Ru纳米颗粒精准锚定于氧空位位点,与相邻不饱和V位点形成Ru-V配位结构,实现了N₂吸附活化和水分子解离供给H的协同催化。DFT计算结合原位XPS揭示了EMSI驱动的定向电荷转移机制和远端路径加氢反应途径。该工作为光催化固氮催化剂的理性设计提供了新思路:**双位点协同策略可有效解决H供给与N₂活化之间的动力学不匹配问题**。未来可通过调控氧空位密度、优化金属载量及探索其他过渡金属(如Os、Ir)进一步拓展该策略的适用性,推动太阳光驱动绿色合成氨技术的发展。
通讯作者简介
孟祥超,中国海洋大学化学化工学院,教授,博士生导师。本科毕业于中国海洋大学,硕士和博士毕业于加拿大渥太华大学。2019年全职加入中国海洋大学。主要研究方向:光电催化裂解海水制氢;光催化/电催化CO2还原、固氮及新型光电催化反应器设计及开发。在光电催化领域发表学术论文60余篇。
李子真,中国海洋大学讲师,硕士生导师,2013年本科毕业于中国海洋大学, 硕士和博士毕业于加拿大渥太华大学(2016、2019),2020年入职中国海洋大学。研究领域主要集中于光催化剂设计及合成、电催化废水处理及回用技术研究。
本文使用的焦耳加热装置是由合肥原位科技有限公司研发,感谢老师支持和认可!
焦耳加热装置
焦耳加热装置是一种新型快速热处理/合成的设备,该设备可使材料在极短(毫秒级/秒级)时间内达到极高的温度(1000~3000℃),升温速率最快可达到10000k/s;通过对材料的极速升温,可考察材料在极端环境、剧烈热震情况下的物性改变,可通过极速升降温制备纳米尺度颗粒,单原子催化剂,高熵合金等。目前广泛应用在电池材料、催化剂、碳材料、陶瓷材料、金属材料、塑料降解、生物质等领域。