专业科学仪器及设备制造商

DOI:10.1021/acs.nanolett.6c01440
全文速览
直接氨燃料电池(DAFC)作为一种碳中和技术前景广阔,但阳极氨氧化反应(AOR)动力学迟缓严重制约其发展。本研究报告了一种碳黑载体上自组装的PtMn纳米簇连续壳层结构,其中Mn以原子级分散形式掺入Pt晶格。通过精细调控Mn含量,实现了位点分化的双位点协同催化机制:Mn位点作为主要活性中心负责NH₃的捕获和脱氢生成NH₂,Pt位点作为次级活性中心吸附额外的NH₂中间体并促进后续N–N偶联。最优催化剂Pt₀.₉₅Mn₀.₀₅‑SACS遵循Sabatier原则,峰值电流密度达22.69 mA·cm⁻²(是商业PtIr/C的9倍),Tafel斜率低至42.86 mV·dec⁻¹以下(原文实际数据:商业PtIr/C Tafel斜率较高,但文中图3e显示Pt₀.₉₅Mn₀.₀₅‑SACS Tafel斜率最小,具体数值见原文补充材料,此处以描述性为主)。在60 ℃组装的DAFC中,该催化剂实现峰值功率密度14.22 mW·cm⁻²,优于多数已报道体系。该工作通过电子结构调控实现位点分化功能,为AOR催化剂设计提供了新思路。
背景介绍
氨作为一种无碳能源载体,具有高能量密度(13.6 MJ·L⁻¹)、易液化和成熟的储运技术,可直接用于直接氨燃料电池(DAFC)发电。然而,阳极氨氧化反应(AOR)涉及多步脱氢和N–N偶联,动力学极其缓慢,是DAFC性能的主要瓶颈。铂(Pt)是目前公认的最佳AOR催化剂,但仍需较高过电位,且易中毒失活。将Pt缩小至纳米簇可最大化原子利用率,而引入第二金属(如Mn)可通过配体效应优化Pt电子结构。Mn具有较低的电负性(1.55 vs. Pt的2.28),易向Pt转移电子,且Mn–N键的形成有望改变反应路径。然而,如何精准构建双金属纳米簇并阐明其协同催化机制仍是挑战。本研究通过自组装策略制备了PtMn纳米簇壳层,揭示了Mn位点与Pt位点的分化功能,实现了高效AOR。
本文亮点
(1)自组装纳米簇壳层结构:PtMn纳米簇(~3 nm)在碳黑表面自组装形成连续壳层(厚~10 nm),原子级分散Mn均匀取代Pt晶格,产生丰富边缘位错和应变场(GPA证实压缩/拉伸应变交替)。
(2)位点分化双位点协同机制:Mn电负性低、d带中心靠近费米能级(–0.209 eV),优先吸附NH₃并脱氢为NH₂;Pt d带中心远离费米能级(–2.019 eV),作为次级位点捕获第二个NH₂,并在Mn–Pt界面促进N–N偶联。理论计算表明,最优Pt₀.₉₅Mn₀.₀₅‑SACS整体能垒最低(0.766 eV)。
(3)卓越性能:峰值电流密度22.69 mA·cm⁻²(商业PtIr/C的9倍),Tafel斜率最低,500圈循环后活性保持89.82%;DAFC中峰值功率密度14.22 mW·cm⁻²(60 ℃),10小时恒流放电保持97.50%。
图文解析
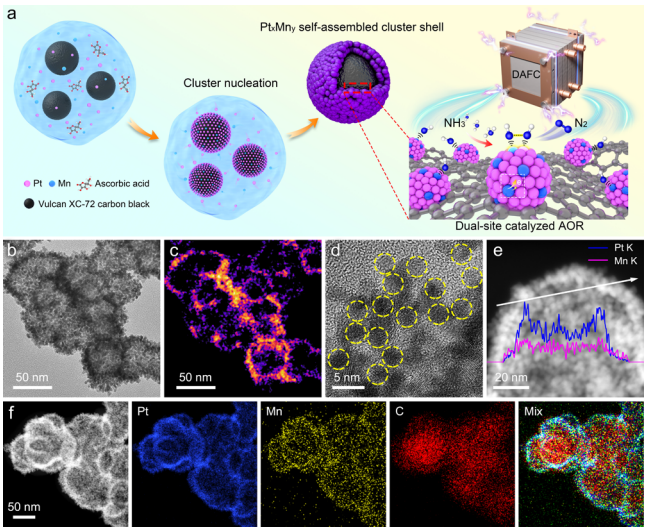
图1:PtₓMnᵧ‑SACS的合成与形貌表征
(a) PtₓMnᵧ‑SACS合成示意图;(b) Pt₀.₉₅Mn₀.₀₅‑SACS的TEM图像,(c) HAADF-STEM图像,(d) AC HAADF-STEM图像;(e) Pt₀.₉₅Mn₀.₀₅‑SACS的EDS线扫描剖面图;(f) STEM-EDS元素面分布图。
图1a展示了以K₂PtCl₄和Mn(NO₃)₂为前驱体,抗坏血酸还原,乙二醇为溶剂的湿化学法制备过程。TEM(图1b)显示碳黑球核被密集的PtMn纳米簇壳层包裹。HAADF-STEM(图1c,d)证实纳米簇尺寸均匀(~3 nm),自组装形成连续壳层。EDS线扫描(图1e)表明壳层厚度约10 nm;元素面分布(图1f)显示Pt(蓝)和Mn(黄)均匀分布于碳核(红)周围,无偏析。
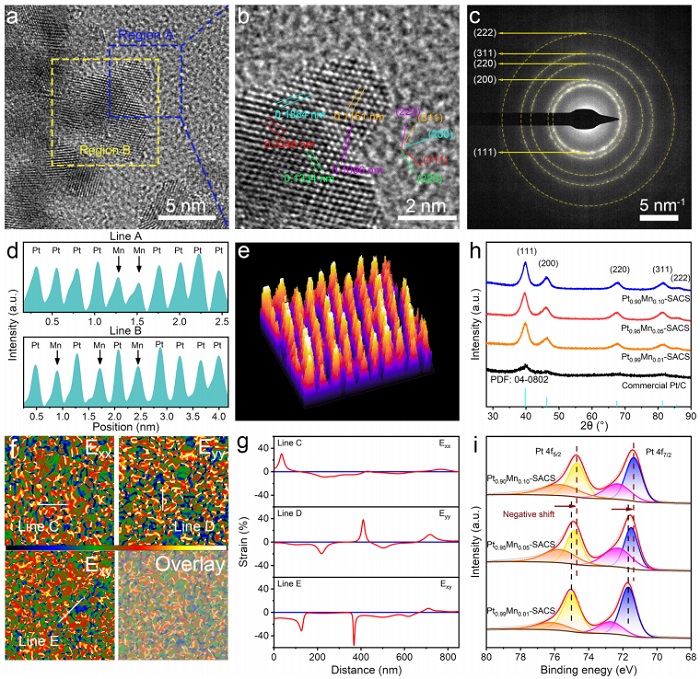
图2:原子结构、晶格应变与电子态
(a) Pt₀.₉₅Mn₀.₀₅‑SACS的AC HAADF-STEM图像;(b) 特定区域的放大图;(c) SAED图像;(d) 图S4中A、B线的亮度强度分布;(e) 选定区域的3D拟合图;(f) GPA分析的应变分量εₓₓ、εᵧᵧ、εₓᵧ等高线图及与AC HAADF-STEM的叠加图;(g) 沿C、D、E线的应变水平;(h) PtₓMnᵧ‑SACS和商业Pt/C的XRD谱;(i) PtₓMnᵧ‑SACS的高分辨Pt 4f XPS谱。
AC HAADF-STEM(图2a,b)显示清晰的(111)、(200)等晶面,晶面间距小于纯Pt(0.2246 nm vs. 0.2265 nm),证实Mn掺入导致晶格收缩。SAED(图2c)呈现多晶环,对应面心立方结构。线扫强度(图2d)和3D拟合(图2e)确认Mn原子随机取代Pt。GPA分析(图2f,g)显示原子尺度压缩/拉伸应变交替,由原子半径差异引起。XRD(图2h)峰位较纯Pt正移,进一步证实晶格收缩。XPS(图2i)中Pt 4f结合能随Mn含量增加而负移,表明Mn向Pt转移电子,Pt富电子化。
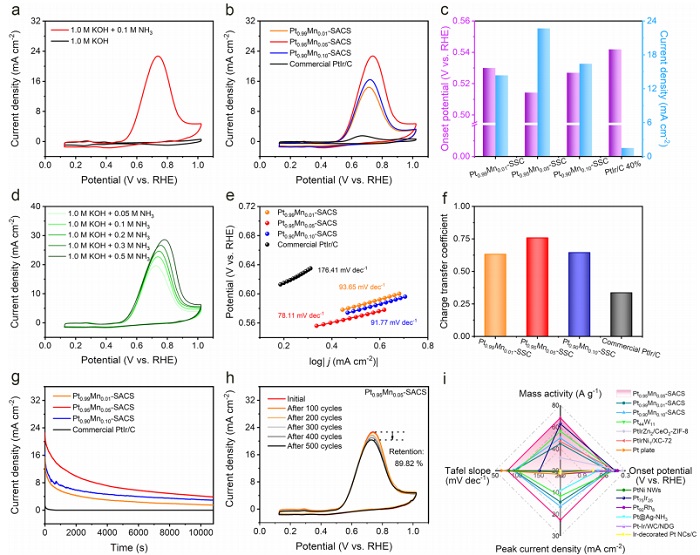
图3:电化学AOR性能
(a) Pt₀.₉₅Mn₀.₀₅‑SACS在有无NH₃时的CV曲线(5 mV·s⁻¹);(b) PtₓMnᵧ‑SACS在Ar饱和1.0 M KOH+0.1 M NH₃中的CV曲线;(c) 起始电位与电流密度;(d) 塔菲尔图;(e) 电荷转移系数;(f) 双电层电容C_dl;(g) 在0.65 V vs. RHE下的计时电流曲线;(h) Pt₀.₉₅Mn₀.₀₅‑SACS循环500次前后的CV曲线;(i) 质量活性与文献对比。
图3a显示加入NH₃后出现明显氧化峰,证实AOR发生。图3b中,Pt₀.₉₅Mn₀.₀₅‑SACS峰值电流密度达22.69 mA·cm⁻²,远超商业PtIr/C(2.45 mA·cm⁻²)。起始电位(图3c)低至0.51 V vs. RHE。塔菲尔斜率(图3d)最小,电荷转移系数(图3e)最大,表明动力学最优。C_dl(图3f)高达8.31 mF·cm⁻²,暴露更多活性位点。计时电流(图3g)显示10800 s后仍保持3.83 mA·cm⁻²,稳定性远优于商业样。500圈循环后(图3h)保留89.82%活性,且ICP-OES未检测到金属溶出。质量活性(图3i)显著优于文献报道。
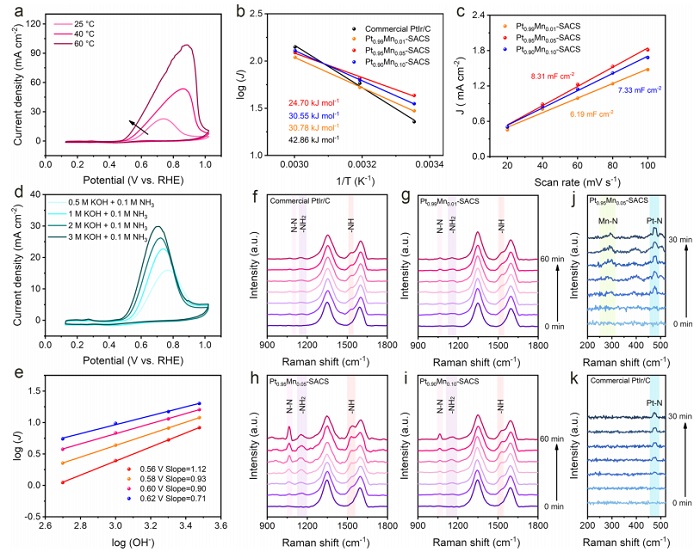
图4:反应动力学与原位拉曼机制
(a) Pt₀.₉₅Mn₀.₀₅‑SACS在不同温度下的CV曲线;(b) Arrhenius图及活化能;(c) C_dl值;(d) 不同KOH浓度下的CV曲线;(e) 电流密度与OH⁻浓度的对数关系;(f-i) 商业Pt/C、Pt₀.₉₉Mn₀.₀₁‑SACS、Pt₀.₉₅Mn₀.₀₅‑SACS和Pt₀.₉₀Mn₀.₁₀‑SACS的原位拉曼谱(氮中间体);(j,k) Pt₀.₉₅Mn₀.₀₅‑SACS和商业Pt/C的原位拉曼谱(金属-N键)。
随温度升高,电流密度增加,Arrhenius拟合得Pt₀.₉₅Mn₀.₀₅‑SACS活化能最低(24.70 kJ·mol⁻¹),证实其本征活性高。OH⁻浓度增加促进AOR,反应级数接近1(图4e),表明OH⁻直接参与脱氢。原位拉曼(图4f-i)检测到*N₂H₄中间体(1063、1153、1535 cm⁻¹),且Pt₀.₉₅Mn₀.₀₅‑SACS峰强最强,证明AOR遵循Gerischer‑Mauerer机制。图4j中观察到Mn–N(~280 cm⁻¹)和Pt–N(~480 cm⁻¹)振动峰,而商业Pt/C仅显示Pt–N(图4k),直接证明Mn和Pt均参与N中间体吸附,实现了双位点协同。
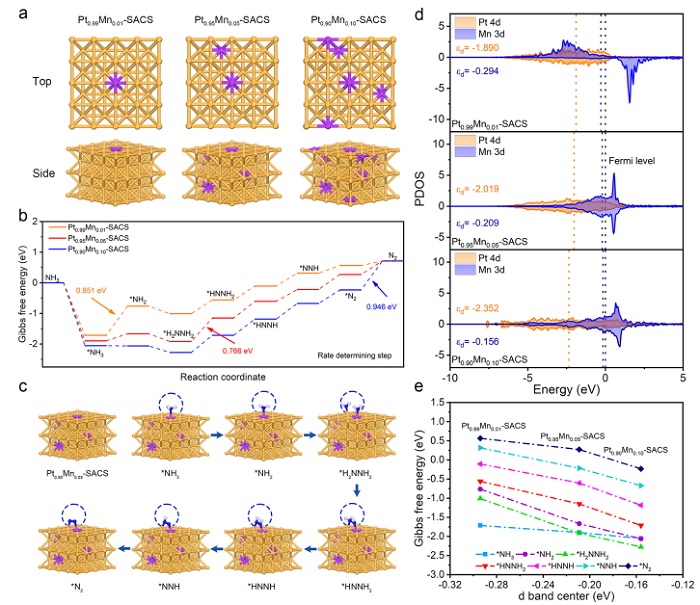
图5:DFT计算与双位点机制
(a) Pt₀.₉₉Mn₀.₀₁‑SACS、Pt₀.₉₅Mn₀.₀₅‑SACS和Pt₀.₉₀Mn₀.₁₀‑SACS的侧视和俯视图(紫:Mn,黄:Pt);(b) AOR的吉布斯自由能图;(c) Pt₀.₉₅Mn₀.₀₅‑SACS上各中间体结构;(d) Pt和Mn的d轨道分波态密度及d带中心位置;(e) d带中心与中间体吸附自由能的关联。
图5a模型基于实验确定的原子比构建。自由能图(图5b)显示:Pt₀.₉₉Mn₀.₀₁‑SACS中NH₃→NH₂为决速步(ΔG=0.951 eV,吸附过弱);Pt₀.₉₀Mn₀.₁₀‑SACS中N₂脱附为决速步(ΔG=0.946 eV,吸附过强);而Pt₀.₉₅Mn₀.₀₅‑SACS整体能垒最低(0.766 eV),符合Sabatier原则。图5c展示双位点路径:NH₃优先在Mn上脱氢为NH₂,Pt捕获第二个NH₂,在Mn–Pt界面发生N–N偶联。d带中心(图5d)表明随Mn含量增加,Pt d带中心远离费米能级(–1.890→–2.352 eV),Mn d带中心靠近费米能级(–0.294→–0.156 eV)。图5e显示吸附强度主要由Mn d带中心决定,最优Mn含量(d带中心–0.209 eV)实现了适度吸附。
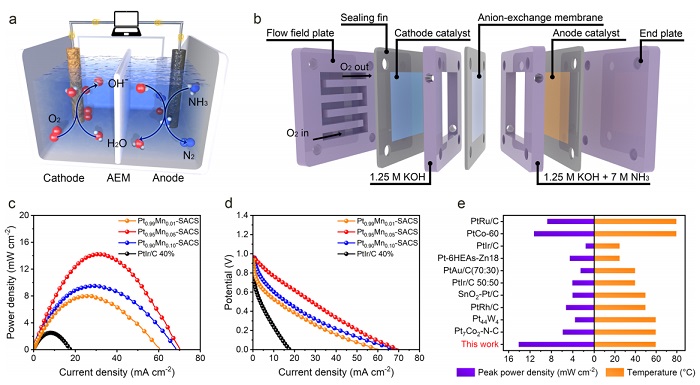
图6:DAFC器件性能
(a) DAFC工作原理示意图;(b) 自组装DAFC的爆炸视图;(c) 不同阳极的极化曲线;(d) 对应的功率密度曲线;(e) 不同催化剂DAFC的峰值功率密度对比。
图6a展示DAFC运行机制:阳极NH₃被OH⁻氧化生成N₂和电子,阴极O₂还原为OH⁻。自组装电池(图6b)采用AEM分隔。图6c中Pt₀.₉₅Mn₀.₀₅‑SACS开路电压0.961 V,功率密度(图6d)峰值为14.22 mW·cm⁻²(60 ℃),远高于Pt₀.₉₉Mn₀.₀₁‑SACS(8.04)、Pt₀.₉₀Mn₀.₁₀‑SACS(9.51)和商业PtIr/C(2.50)。10小时恒流放电保持97.50%电流密度。图6e与文献对比,该器件性能优于多数报道体系,展示了实际应用潜力。
总结与展望
本研究通过湿化学自组装方法成功制备了碳黑负载的PtMn纳米簇壳层催化剂,实现了原子级分散的Mn取代Pt晶格。最优组分的Pt₀.₉₅Mn₀.₀₅‑SACS展现出卓越的氨氧化反应性能,其核心在于Mn和Pt位点的电子结构分化:Mn较低的电负性和靠近费米能级的d带中心使其优先吸附并活化NH₃,而Pt富电子状态和远离费米能级的d带中心则有利于N–N偶联。这种双位点协同机制遵循Sabatier原则,以适中的中间体结合能降低了反应能垒。该工作不仅为AOR催化剂设计提供了“位点分化”新思路,也为DAFC的实用化发展奠定了基础。未来可通过调控纳米簇尺寸、壳层厚度及探索其他低电负性金属(如Cr、V等)进一步优化性能。
通讯作者简介
钱涛,南通大学化学化工学院教授,博士生导师,南通市绿色氢氨储能重点实验室主任。 长期从事电化学储能与催化研究,聚焦氢/氨能源转化、燃料电池、电合成等领域。以通讯作者在 Adv. Mater.、Adv. Funct. Mater.、Nat. Commun.、Nano Lett. 等期刊发表论文多篇,主持国家自然科学基金及省部级项目多项。入选江苏省“333高层次人才培养工程”培养对象。
刘思鸶,南通大学化学化工学院特聘教授。研究方向为电催化、燃料电池、纳米催化材料。在 Nano Lett.、ACS Nano、Adv. Funct. Mater.、Nat. Commun.、Nano Lett. 等期刊发表论文多篇,主持国家自然科学基金青年项目及江苏省自然科学基金。担任南通市绿色氢氨储能重点实验室核心成员。
葛明,南通大学化学化工学院副院长。主要研究领域为电催化、氨氧化、燃料电池催化剂设计。在 Nano Lett.、Chem. Eng. J. 等期刊发表论文多篇,参与国家自然科学基金及产学研合作项目。
本文使用的原位电化学拉曼反应池由合肥原位科技有限公司研发,感谢老师支持和认可!

产品特点:
① 主池体材质是 PEEK/PMMA/PTFE/PI;
② 工作电极为玻碳电极等盘状电极;
③ 参比电极可选银/氯化银电极、硫酸亚汞电极、汞/氧化汞电极和饱和甘汞电极;
④ 对电极可选铂丝环电极和石墨棒电极;
⑤ 可通气氛及电解液静态、动态的研究,整个池体密封性良好;
⑥ 阴极室与阳极室用离子膜(用户自备)隔开,有效避免两极区可能发生的化学反应;
⑦ 光学窗口可选石英、蓝宝石和氮化物窗口,窗口直径 36mm;
⑧ 工作电极表面距光窗距离小于 6mm。