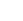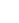专业科学仪器及设备制造商

DOI:10.1039/d5gc07050c
全文速览
烯烃官能团化是合成高附加值化学品的重要途径,但传统方法常依赖化学氧化剂或强酸,且多组分体系中反应路径难以控制。本研究报告了一种非对称电解质电化学策略,通过设计H型电解池中阳极与阴极的溶剂组成差异(阳极无水乙腈、阴极含计量水),精准调控环己烯电氧化生成的碳正离子中间体的后续反应路径。在传统对称电解质中,碳正离子优先与水反应生成氧化产物(2-环己烯-1-醇、2-环己烯-1-酮等),酰胺选择性仅64%。而在非对称体系中,阳极贫水环境迫使碳正离子与乙腈反应生成腈鎓离子,后者扩散至阴极与OH⁻反应,实现酰胺选择性高达98%(Ritter型反应),同时整体法拉第效率达71%。该策略还可实现完全氧化路径(100%选择性)。机理研究结合原位DRIFTS、动力学分析和DFT计算,揭示了外球电子转移生成碳正离子、以及阴极碱性微环境加速酰胺化的关键步骤。该工作为烯烃选择性电转化提供了绿色、可持续的新平台。
背景介绍
烯烃的直接官能团化可高效构建烯丙醇、烯酮、烯丙胺等重要结构单元,广泛应用于医药、材料和精细化工领域。传统方法依赖化学计量氧化剂(如Cr(VI)、Mn(VII))或强酸催化的Ritter反应,存在废弃物多、条件苛刻等问题。电化学合成利用电子作为清洁氧化剂,可在温和条件下生成高活性碳正离子中间体,为烯烃官能团化提供了绿色替代方案。然而,碳正离子反应活性极高,在混合溶剂(如水/乙腈)中易发生不可控的亲核进攻,导致氧化与酰胺化路径竞争,选择性难以调控。现有电化学体系通常只能实现有限选择性(酰胺<64%)。如何通过电极微环境设计解耦竞争路径,实现选择性切换,是该领域的核心挑战。
本研究以环己烯为模型底物,发现碳纸电极能高效介导外球电子转移生成环己烯基碳正离子。通过构建非对称H型电解池(阳极无水乙腈、阴极含水乙腈),利用阳离子交换膜维持电荷平衡,实现了阳极生成腈鎓离子、阴极完成酰胺化的空间解耦,将酰胺选择性提升至98%,并可通过调节阳极水含量一键切换至完全氧化路径。该工作为电化学碳正离子控制提供了新原理。
本文亮点
(1)非对称电解质设计:H型电解池中阳极无水乙腈、阴极含计量水,利用阳离子交换膜分隔,在不破坏整体电荷平衡下创造局部溶剂和亲核性不对称,首次实现了电化学Ritter型酰胺化选择性高达98%。
(2)选择性可切换:通过调节阳极水含量,可实现氧化产物100%选择性或酰胺98%选择性。
(3)碳正离子中间体直接观测:原位DRIFTS在1580 cm⁻¹捕获到环己烯基碳正离子的C=C特征峰,结合DFT计算证实外球电子转移机制。
(4)机理揭示:阴极区产生的OH⁻显著加速腈鎓离子的水进攻,而传统均相体系中水优先攻击碳正离子,非对称设计巧妙利用了阴阳极微环境差异。
(5)底物拓展:适用于多种环状烯烃,但对直链烯烃和缺α-H烯烃无活性,明确了反应适用范围。
图文解析
图1:电催化环己烯氧化(ECOR)性能与机理
(a) 在0.1 M NaClO₄、CH₃CN/H₂O=9/1(v/v)电解质中,环己烯电氧化的产物;(b) 1.8 V(vs. Ag/AgCl)下总产率和法拉第效率随电量的关系;(c) 不同电位下各产物的法拉第效率(200 C电荷);(d) 1.8 V下各产物产率及环己烯转化率随时间的变化;(e) 氧化电流对环己烯浓度的对数依赖关系;(f) 氧化电流对水浓度的对数依赖关系;(g) 活化能平方根与施加电位的线性拟合;(h) 不同电位下碳纸阳极的原位DRIFTS谱图;(i) 环己烯和环己烯基碳正离子的结构及理论模拟红外吸收信号;(j) 不同CH₃CN/H₂O比例下的产物分布。
图1a显示主要氧化产物为醇(1)、酮(2)、环戊烷甲醛(3)和环己酮(4)。图1b表明法拉第效率达60–75%。图1d显示醇(1)随时间逐渐转化为酮(2),而环收缩产物(3,4)一旦生成便稳定。图1e-f确定反应对CHX为一级、对水为0.44级,图1g中E_a^(1/2)与E线性相关(R²=0.965),证明确实为外球电子转移。原位DRIFTS(图1h)在>1.4 V时于1580 cm⁻¹出现新峰,与图1i中理论模拟的碳正离子C=C振动一致,直接证明了碳正离子中间体的存在。图1j显示当水含量降至<2.5 vol%时,开始出现酰胺产物5。
图2:反应网络示意图
该图总结了反应机理:环己烯(CHX)在碳纸电极上经外球电子转移失去一个电子,生成碳正离子中间体I和II。在高水含量下,中间体II被水亲核进攻得到醇(1),进一步氧化为酮(2);中间体I可脱质子生成环己酮(3)或重排后水进攻生成环戊烷甲醛(4)。在低水含量下,中间体II被乙腈捕获生成腈鎓离子III,后者与水反应生成酰胺产物5。该网络清晰解释了产物分布与溶剂组成的依赖关系。
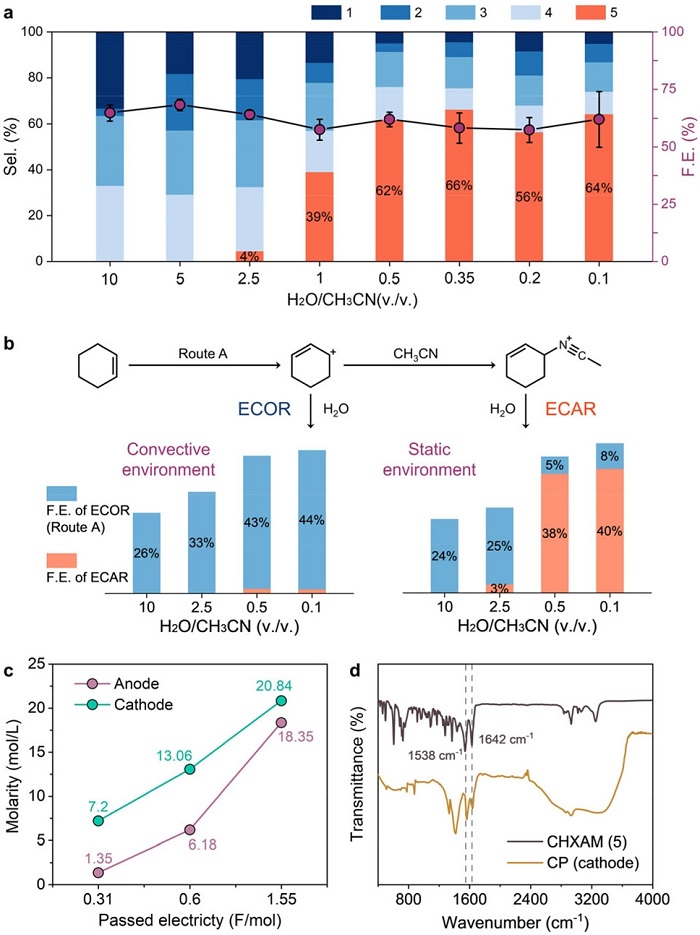
图3:电极微环境对ECOR和ECAR动力学的影响
(a) 不同水/乙腈比例下的产物选择性;(b) 静置和600 rpm搅拌下的单电解池反应;(c) 装有Nafion 117膜的H型电解池中阳极区和阴极区酰胺产物浓度的变化;(d) 电解后阴极电极的FTIR谱图。
图3a显示酰胺选择性随水含量降低而升高,但低于0.5 vol%后不再增加(~60%)。图3b表明在低水含量(ECAR显著)时,搅拌几乎完全抑制酰胺生成,转而促进氧化产物1,说明酰胺化对对流高度敏感。图3c显示H型电解池中阴极区酰胺浓度高于阳极区。图3d的FTIR在阴极沉积物中检测到酰胺的特征峰(1538和1642 cm⁻¹),红移归因于氢键聚合,证实酰胺在阴极富集。
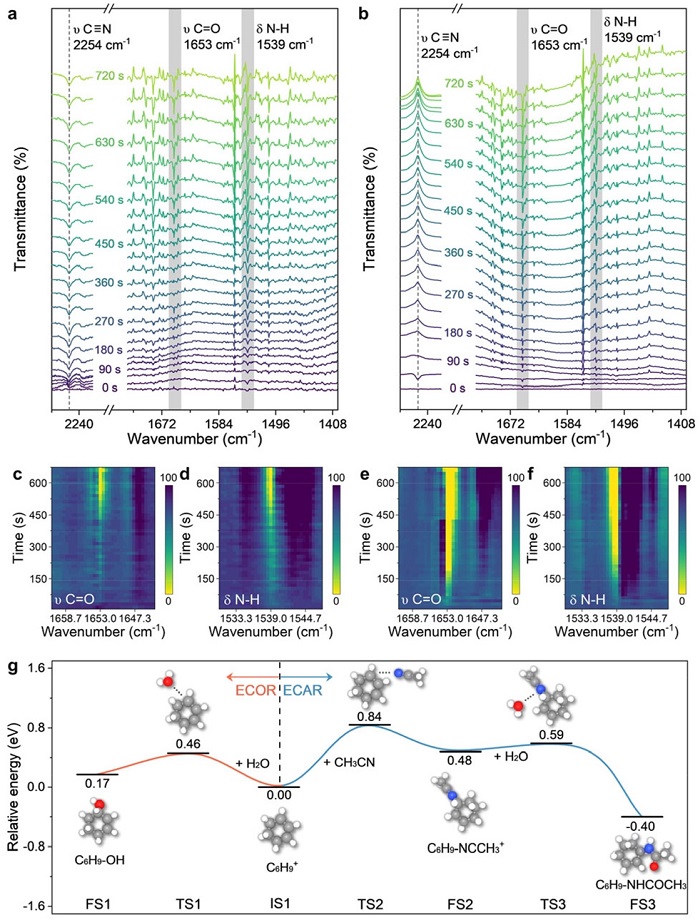
图4: 原位DRIFTS与DFT计算
在0.1 M NaClO₄、H₂O/CH₃CN=1/200电解质中,1.7 V(vs. Ag/AgCl)下:(a,b) 阳极和阴极的时间分辨原位DRIFTS谱图(以加入CHX后、施加电位前的谱图为基线);(c-f)阳极和阴极处酰胺特征峰的时间演化,色标表示相对透射率;(g) ECOR和ECAR中涉及碳正离子关键步骤的DFT能垒计算。
图4a,b显示阳极和阴极均出现酰胺的C=O(1653 cm⁻¹)和N-H(1539 cm⁻¹)特征峰。图4c-f中阴极信号出现略早于阳极,且乙腈的C≡N峰在阴极处减弱(消耗),在阳极处增强(水消耗导致乙腈富集)。DFT(图4g)计算表明:碳正离子与水反应能垒(0.46 eV)低于与乙腈反应(0.84 eV),解释了高水含量时氧化占优;而腈鎓离子与水反应能垒仅0.11 eV,远低于碳正离子与水的0.46 eV,因此一旦生成腈鎓离子,在阴极碱性环境中极易转化为酰胺。
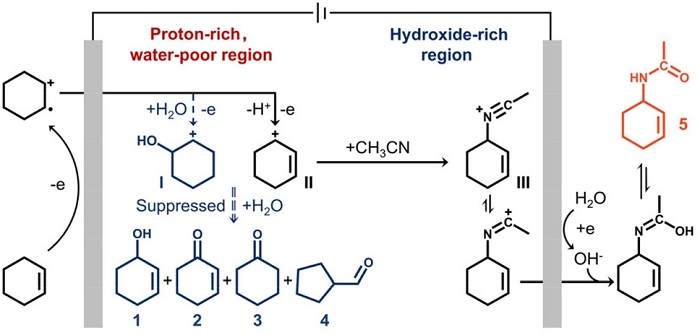
图5:非对称电解池原理与性能对比
(a) 非对称电解质电解池实现高选择性ECAR过程,插图为阳极室溶液呈浅红色、阴极室无色;(b) 普通均相电解质系统只能实现有限的ECAR选择性。非对称体系中阳极室溶剂为无水乙腈,阴极室为乙腈与化学计量水的混合物;对称体系中两室均为含0.1 vol%水的乙腈。
非对称设计将酰胺选择性从64%提升至98%,法拉第效率从64%升至71%(图5b)。阳极溶液呈红色归因于生成的腈鎓离子中间体。该设计解耦了碳正离子生成与酰胺化步骤:阳极贫水迫使碳正离子与乙腈反应生成腈鎓离子,后者在电场下迁移至阴极,与阴极产生的OH⁻快速反应生成酰胺。若阳极含水>2.5 vol%,则切换至完全氧化路径(100%选择性)。
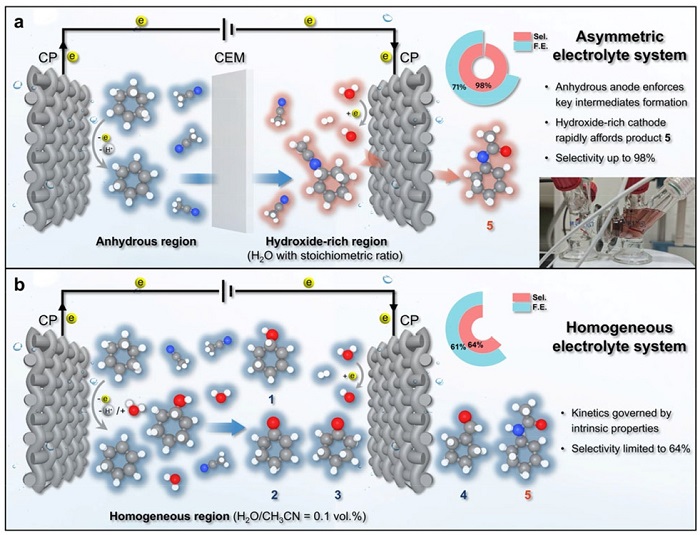
图6:底物拓展与长期稳定性
甲基环戊烯和环辛烯表现出类似的电氧化和酰胺化活性,而1-己烯(直链)和苯乙烯(缺α-H)无响应。DFT计算表明直链烯烃电子转移能垒高,苯乙烯无法生成稳定碳正离子。长期电解实验中,底物消耗后电流下降,补充CHX和水后电流完全恢复,且可多次循环,表明体系具有良好的稳健性和可再生性。
总结与展望
本研究开发了一种非对称电解质电化学平台,通过对H型电解池中阴阳极溶剂组成的差异化设计,精确控制烯烃衍生的碳正离子中间体的反应路径,实现了氧化与酰胺化反应的高选择性切换。以环己烯为模型,氧化路径可获100%烯丙醇/烯酮产物,酰胺路径选择性达98%。核心创新在于:阳极无水环境迫使碳正离子与乙腈生成腈鎓离子,阴极碱性环境加速其水解脱除,二者协同打破了均相体系中水优先亲核的限制。原位DRIFTS直接观测到碳正离子中间体,DFT计算验证了能垒差异。该策略仅以水为氧源、乙腈为溶剂兼亲核试剂,无需外加氧化剂或强酸,体现了绿色化学原则。
该工作为电化学碳正离子调控提供了新范式,并可拓展至其他环状烯烃。未来可结合流动电化学放大、使用可再生能源电力,进一步推动烯烃电转化技术的绿色化和实用化。
通讯作者简介
丁维平,南京大学化学化工学院教授,博士生导师。1983年获南京大学化学系物理化学专业学士学位,1992年获该校博士学位。1983年至1986年任教于扬州大学,1993年至1996年在南京大学物理系从事博士后研究,1999年至2000年任美国加州大学伯克利分校化工系访问学者,2004年至2005年任哈佛大学化学系访问学者 。2007年任中国石化上海石油化工研究院-南京大学催化材料与技术联合实验室主任,2008年至2011年任南京大学化学化工学院副院长。长期从事催化材料与催化化学研究,在多相催化、分子筛催化、低碳转化等领域取得系列创新成果。以通讯作者在 Nat. Commun.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、Appl. Catal. B 等期刊发表论文200余篇,主持国家自然科学基金重点项目、科技部重点研发计划等多项国家级项目。
本文使用的原位红外漫反射测试由合肥原位科技有限公司提供,感谢老师支持和认可!